Development News Brief
Get Galaxy

new: % hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist
upgrade: % hg pull -u -r 17d57db9a7c0
# EMBOSS Tool Migration
The Emboss 5.0.0 tools, datatypes and functional test data files have been removed from the Galaxy code base in this revision and have been migrated to the Galaxy tool shed. This will affect you only if you are using the Emboss 5.0.0 tools in your local Galaxy environment by having one or more of them defined in you local tool_conf.xml file.
**After you update your Galaxy code base to this revision (if you use any of the [EMBOSS](http://emboss.sourceforge.net) tools), attempting to start your Galaxy server will display a message with instructions on what to do to install the tools from the tool shed.** You can choose to not install the tools and start your Galaxy server - it will start on this second attempt.

If you decide to install the tools, the location in which the Emboss repositories will be installed is the value of the "tool_path" attribute in in the <tool> tag of the file named "migrated_tools_conf.xml" in your Galaxy installation directory. The default location setting is "../shed_tools", which may be problematic for some cluster environments, so make sure to change it before you execute the installation process if necessary.
If your local Galaxy environment is configured to run multiple web front-ends, you should start up a single front-end for your Galaxy server and watch its paster log to see the instructions. If you start all configured web front-ends, only the first one started will produce the instructional message, and others will start up your Galaxy server.
At any point, you can install the EMBOSS tools by executing the following from your Galaxy installation directory.
sh ./scripts/migrate_tools/0002_tools.shAfter installation, the tools should be displayed in your Galaxy tool panel in precisely the same way that they were displayed before the installation. In order for this to happen, however, you must have the tools defined in your local tool_conf.xml file as you want them displayed in your tool panel before you perform the installation. After the installation, you can remove the entries for them from your tool_conf.xml file.
We highly recommend that you read the following sections of the Galaxy tool shed wiki (if you have not already done so) to get all of the details about how this process works.
Migrating tools from the Galaxy distribution to the Galaxy Main tool shed
# New Tool Integration Testing
Use Galaxy’s functional test framework to test tools installed into your local Galaxy instance from Tool Shed repositories that contain tools with defined functional tests. For tests to work, the repository must include a directory named test-data somewhere in its file system hierarchy, and all functional test input and output datasets must be included in this directory. Read more…
# Updates to Tool Panel Configuration
Galaxy now supports multiple tool panel XML configuration files for populating the tool panel. You can have any number of file names defined in the comma-separated list of tool panel configuration files for the "tool_config_files" setting in your universe_wsgi.ini file.
For example:
tool_config_files = tool_conf.xml,tool_conf1.xml,shed_tool_conf.xml,shed_tool_conf1.xml, etc.
Managing the layout of your Galaxy tool panel is easier! New tool panel items manually added to a tool panel configuration file will now be inserted into an existing integrated_tool_panel.xml file in the proper location. Read more…
# Multiprocess Job Handling
Galaxy has previously been able to split into multiple processes to better spread the load of handling web traffic, but only one process could be used to handle job management. Galaxy can now be split in to any number of web processes, and any number of job handlers. A single job manager must be run to assign jobs to handlers. If you’ve configured Galaxy with multiple processes as per the “Production Server” documentation, configuration changes will be necessary to use this new functionality. If you do not change your configuration, each web processes will manage its own jobs, which you almost certainly do not want. Read more…
# UCSC Display Sites
If use_remote_user = True in your config, Galaxy previously used a hardcoded list to determine what servers were allowed to bypass security to read datasets for display. This list is now configurable via the display_servers. If you have modified galaxy-dist/lib/galaxy/web/framework/middleware/remoteuser.py to modify the list of hardcoded servers you will most likely experience merge conflicts upon pulling this distribution. If this is the case, you should remove your local modifications and use the display_servers configuration option, which is documented in universe_wsgi.ini.sample.
# Enhanced OpenID Support
Abstract OpenID providers to be defined outside of the Python code and to allow customization of post-authentication actions (currently restricted to storing sreg attributes into user preferences; but more functions can be added as needed). See individual examples in openid/ and the list of enabled OpenID providers in openid_conf.xml.sample to add your own.
# New Configurable Tool Output Location
Tools using the process described at ‘Admin/Tools/MultipleOutputFiles’ to write multiple output files to the temporary directory defined as new_file_path may now write to the job working directory instead. This can have a performance benefit if there are a lot of files in the directory set in new_file_path. A configuration option, collect_output_from has been added that controls where Galaxy will look for these outputs. By default, it will look in both places since many of the tools in the Galaxy distribution use new_file_path.
# Tools
Admin/Config/Tool Dependencies
- NGS: GATK Tools (beta)
- Added GATK version 1.4
- Compiled from v1.4-18-g80a4ce0
- Genome Analysis Toolkit (beta) tool group includes: Depth of Coverage on BAM files; Print Reads from BAM files; Realigner Target; Creator for use in local realignment; Indel Realigner - perform local realignment; Count Covariates on BAM files; Table Recalibration on BAM files; Analyze Covariates - draw plots; Unified Genotyper SNP and indel caller; Variant Annotator; Variant Filtration on VCF files; Select Variants from VCF files; Variant Recalibrator; Apply Variant Recalibration; Validate Variants; Eval Variants; Combine Variants.
- Additional tool documentation developing over next few months. For now, refer to GATK tool author help.
- Added GATK version 1.4
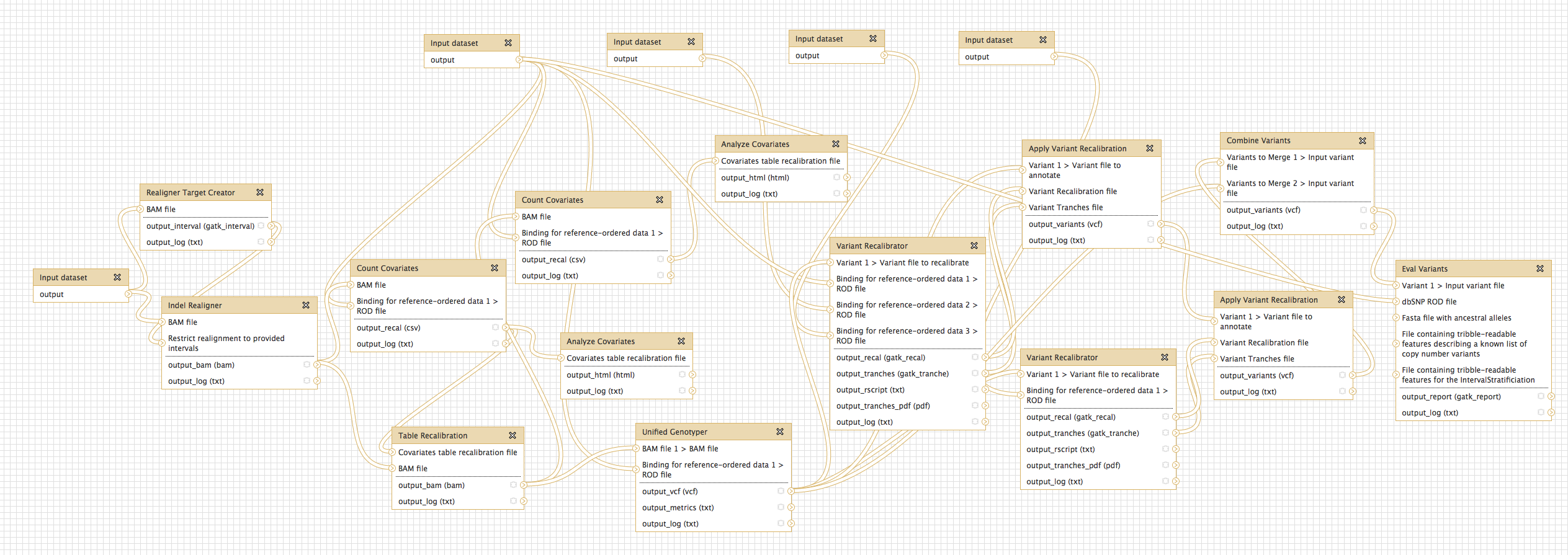 * **NGS: Variant Detection**
* Added **[FreeBayes](http://github.com/ekg/freebayes)** latest version (a46483351fd0196637614121868fb5c386612b55)
* Requires **[SAM Tools](http://samtools.sourceforge.net/), version 0.1.16-0.1.18** *strongly recommended*
* **NGS: Mapping**
* Correction made for **[BWA](http://bio-bwa.sourceforge.net/) -R** option.
* Updated **Megablast** now uses NCBI BLAST+ (changeset [0b5cb60e4810](https://bitbucket.org/galaxy/galaxy-central/changeset/0b5cb60e4810#chg-tools/metag_tools/megablast_wrapper.xml))
* **[BLASTN](http://blast.ncbi.nlm.nih.gov/Blast.cgi) version 2.2.25**
* Simplified indexing. Obtain from [NCBI](ftp://ftp.ncbi.nlm.nih.gov/blast/db/) or use [formatdb](ftp://ftp.ncbi.nih.gov/blast/documents/formatdb.html) to create your own. [Read more...](/admin/NGS Local Setup/)
* Updated **Parse blast XML output**
* Improvements to merging blast xml - `peterjc/galaxy-central/split_blast2` (pull request #41)
* **NGS: [SAM Tools](http://samtools.sourceforge.net/)**
* Have implicit `SAM to BAM` converter sort the output `BAM` file so that indexing will not fail.
* **NGS: RNA-seq**
* Updated **[Trinity](http://sourceforge.net/projects/trinityrnaseq/files/)** wrapper (alpha).
* Trinity performs de novo assembly of RNA-Seq data. [Read more...](http://trinityrnaseq.sourceforge.net)
* Wrapper supports **Trinity r2012-04-22-beta** (and perhaps a few versions earlier).
* All commands included, **Inchworm, Chrysalis, and Butterfly**, see `/tools/ngs_rna/trinity_all.xml`.
* Updated **[Cuffdiff](http://cufflinks.cbcb.umd.edu/manual.html)**
* Galaxy tool form default for `min-alignment-count` is now the same as line command default.
* **Get Data**
* Update to use **[WormBase 2](http://www.wormbase.org)**.
* Correct issues with **[HbVar](http://globin.bx.psu.edu/hbvar/menu.html)**.
* **Display Applications**
* Added **[Integrated Genome Browser (IGB)](http://bioviz.org/igb/index.html)** as a new display option.
* **NGS: Variant Detection**
* Added **[FreeBayes](http://github.com/ekg/freebayes)** latest version (a46483351fd0196637614121868fb5c386612b55)
* Requires **[SAM Tools](http://samtools.sourceforge.net/), version 0.1.16-0.1.18** *strongly recommended*
* **NGS: Mapping**
* Correction made for **[BWA](http://bio-bwa.sourceforge.net/) -R** option.
* Updated **Megablast** now uses NCBI BLAST+ (changeset [0b5cb60e4810](https://bitbucket.org/galaxy/galaxy-central/changeset/0b5cb60e4810#chg-tools/metag_tools/megablast_wrapper.xml))
* **[BLASTN](http://blast.ncbi.nlm.nih.gov/Blast.cgi) version 2.2.25**
* Simplified indexing. Obtain from [NCBI](ftp://ftp.ncbi.nlm.nih.gov/blast/db/) or use [formatdb](ftp://ftp.ncbi.nih.gov/blast/documents/formatdb.html) to create your own. [Read more...](/admin/NGS Local Setup/)
* Updated **Parse blast XML output**
* Improvements to merging blast xml - `peterjc/galaxy-central/split_blast2` (pull request #41)
* **NGS: [SAM Tools](http://samtools.sourceforge.net/)**
* Have implicit `SAM to BAM` converter sort the output `BAM` file so that indexing will not fail.
* **NGS: RNA-seq**
* Updated **[Trinity](http://sourceforge.net/projects/trinityrnaseq/files/)** wrapper (alpha).
* Trinity performs de novo assembly of RNA-Seq data. [Read more...](http://trinityrnaseq.sourceforge.net)
* Wrapper supports **Trinity r2012-04-22-beta** (and perhaps a few versions earlier).
* All commands included, **Inchworm, Chrysalis, and Butterfly**, see `/tools/ngs_rna/trinity_all.xml`.
* Updated **[Cuffdiff](http://cufflinks.cbcb.umd.edu/manual.html)**
* Galaxy tool form default for `min-alignment-count` is now the same as line command default.
* **Get Data**
* Update to use **[WormBase 2](http://www.wormbase.org)**.
* Correct issues with **[HbVar](http://globin.bx.psu.edu/hbvar/menu.html)**.
* **Display Applications**
* Added **[Integrated Genome Browser (IGB)](http://bioviz.org/igb/index.html)** as a new display option.
# Tool Shed
- Features
- The tool shed’s upload process will automatically eliminate undesired directories (.svn, .git, .hg, .cvs) and file (hgrc, .jhg_archival.txt, .DS_Store) from the archive before committing the contents to the repository. This provides for a more pristine environment for those that install the repository locally.
- You can now browse your invalid tools in the tool shed, and clicking on an invalid tools will display information about why it is invalid. The definition of a valid tool in the tool shed has always been restricted to the tool properly loading in a Galaxy instance and that no required dependent files are missing. If a tool is not valid, it will not be returned in a search and it cannot be automatically installed (unless it belongs to a repository containing other valid tools). Only valid tools should be shared in the tool shed or its value will diminish over time. Tool developers should browse their invalid tools and make corrections to them if necessary.
- Reminder to include tool dependency installation instructions or licensing information in your repository (wiki). Tool dependency installation instructions or licensing information should be included in your repository in a file named one of (case is ignored)
readme, readme.txt, read_me, read_me.txt. When a user installs a single repository into their local Galaxy instance, the contents of the file will be displayed on the tool panel section selection page. Care must be taken when including instructions or other information inREADMEfiles to ensure the content is clear and correct.
- Updates:
- Mercurial eggs have been introduced to handle all tool shed mercurial-related features, so mercurial package installation is no longer necessary to use your local Galaxy instance in combination with any Galaxy tool shed. However, you’re still required to install mercurial in order to clone any of the Galaxy / Galaxy tool shed source code repositories ( galaxy-central, galaxy-dist, etc ) and get source code updates for them using mercurial.
- Invalid tools are now displayed in the “Preview tools and inspect metadata by tool version” section when viewing or managing the repository.
- Enable a tool shed administrator to reset all repository metadata in the entire tool shed.
- Enable a tool shed admin to be able to view and regenerate tool shed statistics.
# Galaxy Track Browser (GTB)
- Enable resizing of all tracks.
- Ensure reference track does not scroll.
- Use hash as location in Trackster.
- Use strand coloring for feature tracks.
- Interval datasets can be visualized.
- Use
tabixfor indexing and queryingGTFfiles (much faster!).
# Workflows
- Added “run workflow” directly to the workflow editor’s menu.
- New workflow importing API script -
jmchilton/galaxy-central-preseeding/import_workflow_api_script(pull request #39) - Added the ability to flag a workflow for the toolbox menu when adding via the API -
jmchilton/galaxy-central-preseeding/workflow_import_menu_api(pull request #40) - Workflow Extraction Fix - Input datasets from nested
group/repeat/conditionalparameters should now be connected correctly in the output workflow. - Workflow Editor fix for the case where
RenameActionshad null action arguments vs being unspecified or {} as expected. This situation exists in very few rename actions and hasn’t been reproduced, may be caused by legacy workflow steps.
# User Interface (UI)
- Updated UI Styling for Menus, Panels, Fonts, and much more. Comments welcome the galaxy-dev@bx.psu.edu mailing list.
- Incremental display for tabular datatypes.
- Significantly improve handling of SAM rendering with the new tabular display, including optional parameters.
- For all tabular display: numerical columns align right, text aligns left.
- Updates to tabular chunked display to force text content of cells. (resolves VCF display issues)
- Galaxy will now warn clients that try to navigate away from the Galaxy analysis interface while a file is uploading. This was likely the cause of many interrupted uploads in the past.
- Model–view–controller (MVC)
- Add
backbone.js, underscore.js, and handlebars.jslibraries in a new/libsdirectory and modify pack scripts accordingly. - Use Backbone-based code to render and search tool panel.
- Add a
transform_publicnamemethod that coerces a provided string into a valid public name. This is now invoked during user creation to help guide the selection of an acceptable public name. - Add
rerunandshow infobuttons to discarded datasets (usually deleted before job finished)
- Add
# Source
- Remove
enable_api flag; API is now enabled by default and cannot be disabled. - Enable dynamic options for
DrillDownSelectToolParameter. - Rework the display of
DrillDownform elements. - Add require_login(=False) and display_interface(=True) flags to tools.
- New config option, and change in default behavior, regarding ‘dataset path display’ in “View Details”. Administrators can now always see the full path, and if the
expose_dataset_path optionisTrue, so can regular users. - Basic functionality was added for accessing the API through a session (if available) instead of an API key.
- File path now shown (when appropriate) when datasets are accessed via the history api. (This was incorporation and slight update to cjav/galaxy-central/expose-dataset-full-path-through-the-api (pull request #43)).
- Add basic tool execution to the API.
# Bug Fixes
# Announcements
GCC2012 Update

The 2012 Galaxy Community Conference (GCC2012) will be held July 25-27, in Chicago. The conference will feature two full days of presentations and discussions, and a whole day of training has been added this year.
Early registration is now open and will close June 11. Early registration is very affordable, especially for post-docs and students.
Training Day: Help Set the Agenda

The GCC2012 Training Day needs your help! Training Day offers 90 minute training sessions in three parallel tracks throughout the day for a total of 12 sessions. We need your input on how to best allocate those sessions to topics. Please take a few minutes to indicate your top choices for what you would like to see covered on the GCC2012 Training Day.
----
# About Galaxy
The GalaxyTeam is a part of BX at Penn State, and the Biology and Mathematics and Computer Science departments at Emory University.
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State, and Emory University.
Join us at Twitter @galaxyproject or just read our tweets Galaxy on Twitter