Development News Brief
Get Galaxy

new: % hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist
upgrade: % hg pull -u -r 40f1816d6857
Important Changes to Tool Organization
The Emboss tools and Emboss datatypes will be eliminated from the Galaxy distribution in the NEXT release (not this one). Other tools currently in the Galaxy distribution will be eliminated in following releases. Those hosting local Galaxy instances should read this revised “Migrating tools” section of the Galaxy tool shed wiki to understand how this process will work:

In 2012, the Galaxy development team will begin the process of migrating the tools that are currently available in the Galaxy distribution to the Galaxy Main tool shed. This will enable those that host local Galaxy instances much more flexibility in choosing to provide only those specific tools in which their users are interested. Read more…
XML configuration files used to populate your Galaxy tool panel
In the past, the file named by your “tool_config_file” configuration setting in your “universe_wsgi.ini” file was the only file used to populate your Galaxy tool panel. The default name for this file is tool_conf.xml. Since this was the only file involved in populating your Galaxy tool panel, it defined the items (tools, workflows, sections and labels) that would be displayed and the way in which they would be arranged. Read more…
Managing the layout of your Galaxy tool panel
The 3 or more files described in the previous section (“tool_conf.xml”, one or more “shed_tool_conf.xml files”, and “migrated_tools_conf.xml”) are all used to load tool panel items (tools, sections, labels and workflows). A new file named integrated_tool_panel.xml has been introduced to define the arrangement for displaying these loaded items in your Galaxy tool panel. Read more…
Galaxy Tool Versions
When included in the Galaxy distribution, tools are defined by “id” and “version”, among other attributes. For example, the filter tool has id=“Filter1” and version=“1.1.0”. When installed from a tool shed, the tool’s id becomes its “guid” attribute from the tool shed. If it is migrated from the Galaxy distribution to the tool shed, the filter tool will have the guid: “toolshed.g2.bx.psu.edu/repos/devteam/filter/Filter1/1.1.0”. To provide backward compatibility for Galaxy workflows and the rerun button in a Galaxy history item, a mapping between the tool’s old id and version and its new id (guid) is provided by building a chain of relationships between tool versions. This happens automatically for every tool that is loaded into your Galaxy instance. Read more…
New tool shed related features in Galaxy
-
When an installed tool shed repository that contains tools is being reinstalled, allow the admin to select a different section in the tool panel (than was originally selected when the repository was last installed) to contain the included tools.
-
Load proprietary datatypes from installed tool shed repositories before the datatypes in the Galaxy distribution are loaded. We do this because the distribution includes some extremely generic sniffers (e.g., text,xml) which will catch pretty much anything, making it impossible for proprietary sniffers to be used. Proprietary datatypes contained in installed repositories are loaded in order of oldest installation first, followed by next oldest installation, etc. In handling conflicts (2 different datatypes that use the same extension), the rule is that a datatype currently being loaded will always override a conflicting datatypes that was previously loaded. Since datatypes defined in Galaxy’s datatypes_conf.xml file are loaded last, they will override conflicts that may occur if tool shed repositories that contained datatypes were installed. If a new tool shed repository is being installed into a running Galaxy instance, conflicting datatypes will not override those currently in the datatypes registry.
Tool shed related fixes in Galaxy
- Fix for rendering the page that allows you to select a tool section in which to include tools contained in a repository installed from a tool shed where the tool panel includes tool section labels.
- Load empty tool sections into the in-memory tool_panel dictionary, just don’t display them in the tool panel.
# New & Updated Tools
Many tools have been recently upgraded. Please review Admin/Config/Tool Dependencies for these and other recently updated Tool Dependencies. Please see * Galaxy’s Main Tool Shed * for additional new tools.
- RNA-Seq Tools
- Added CuffMerge version 1.0.0
- Requires helper script: gtf_to_sam version 1.3.0
- Updates for Cufflinks/compare/merge/diff tools
- Modified default parameter values on tool form
- Improved error message when bias correction/sequence data cannot be used
- Added CuffMerge version 1.0.0
- Updates for TopHat
- Remove maximum value for TopHat parameter
initial_read_mismatches
- Remove maximum value for TopHat parameter
- Added RViewer external display application
- Updated IGV external display application, so that displays using vcf_bgzip will now maintain vcf headers
Galaxy Track Browser (GTB)
- New:
- Enable visualization of ENCODE peak tracks (see graphic below)
- Dynamic filtering of read tracks using quality scores
- Enable toggling between groups of individual tracks and composite tracks
- Enable Composite Tracks to be saved and restored
- Make track min and max values editable inline
- Save and restore track/group filters and tool state
- Bug fixes:
- Clear reference track when changing chromosomes
- To only show differences if reference data is available
- Use sum rather than mean for data aggregation in BBIDataProvider
- Indicate changes when config values are changed or items are reordered or grouped
- Move ‘more rows’ icon from tile level to track level
Trackster visualization of ENCODE and Composite Track data using dynamic filters
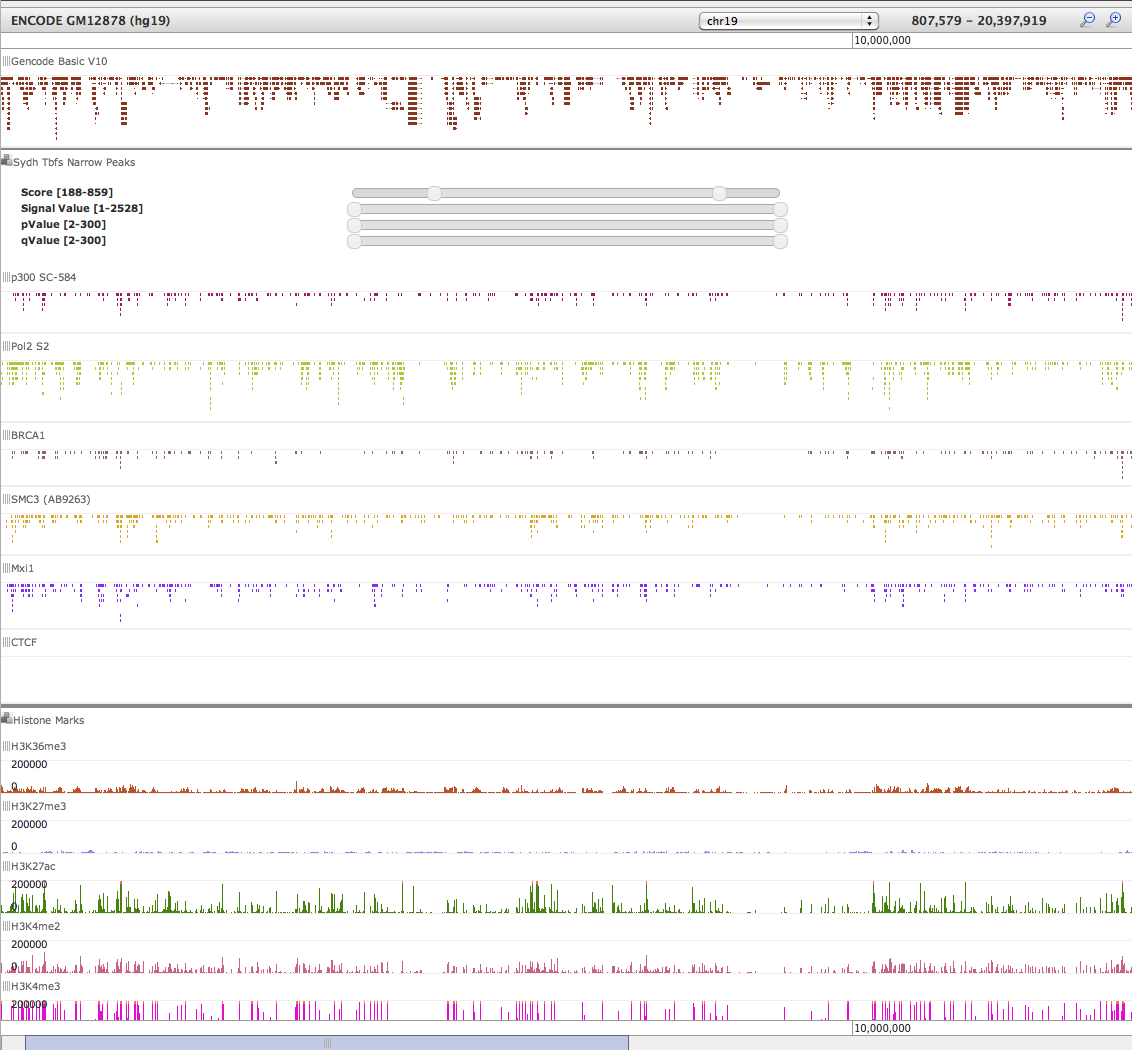
# Workflows
- Properly select only Input Dataset steps for multi-input configuration
- Update and clarify label selector
- Multiple run workflows now copy input datasets to newly created histories when used.
- Add a more useful error message when input keys don’t map upon executing a workflow instead of tossing to Server Error:
- example, This happens in situations where outputs are conditionally filtered yet still used in the workflows. (See SICER’s significant_islands_summary_output_file)
- Add the ability to remove workflows that have been shared with you
- When workflow output is placed into separate new histories, naming is now based on the varied input dataset instead of the execution number:
- example, ”
on <varied_input>”.
- example, ”
- Add the ability to view your own workflows in plain display mode as if you had shared it
- Additional rename options available to Rename Datasets action (thanks to Dave Walton)
Syntax:
#{inputs_file_variable | option 1 | option n}
where:
input_file_variable = is the name of a module input variable
| = the delimiter for added options. Optional if no options.
options = basename, upper, lower
basename = keep all of the file name except the extension
(everything before the final ".")
upper = force the file name to upper case
lower = force the file name to lower case
Example:
#{input1 | basename | upper}User Interface (UI)
- Improved language in Import workflow dialog box
- Change default sort order for Shared Data -> Pages list to be by update time
- Style updates for embedded items to reduce visual footprint
Data and Libraries
- In library uploads:
- Allow inheriting the existing datasets metadata when replacing
- Allow setting library metadata when adding datasets from a history
CloudMan
- A larger tools volume (10GB vs old 2GB) is now the default for any new CloudMan cluster making it easier to customize your Galaxy Cloud instances
- A preliminary support for OpenNebula cloud type exists within CloudMan (thanks to Mattias de Hollander)
- Please continue to use AMI ami-da58aab3 for Galaxy Cloud clusters, as listed on usegalaxy.org/cloud. There is another AMI dated from Feb 26, 2012 that was not created by the Galaxy Team and is not supported by us.
Source
- Parameterize per-tool job runners so that parameter name/value pairs can be used to define multiple runners per tool
- Documentation is in universe_wsgi.ini.sample
- Add ‘params’ column to jobs table to store job parameters
- Add source parameter for all jobs initiated in Trackster
- Enable Pages and Trackster by default in universe_wsgi.ini.sample
- History export now possible for a guest user
- Egg Update: boto 2.2.2
- Preliminary autopacking for javascript:
- Disabled by default (
pack_scriptsin universe_wsgi.ini.sample) and the packed scripts are still in the distribution, to be removed at some point.
- Disabled by default (
- Job Splitting:
- Merged in
peterjc/galaxy-central/split_blast(pull request #37). Provides support for running parallelized blast, merging xml, and some refactoring.
- Merged in
- New universe_wsgi.ini.sample configuration option (
sanitize_all_html):- Enabled by default; to prevent html dataset display sanitization, set to false
- DRMAA Job Runner changes:
- Set a descriptive job name in the DRM
- Use the value of
cluster_files_directoryinstead of the hardcodeddatabase/pbs/directory
Bug Fixes
Announcements
March 2012 Galaxy Update
GCC2012 Update


Galaxy is Hiring!
Want to work on one of the fastest growing open source bioinformatics projects around? The [Galaxy Project](http://galaxyproject.org/), a highly successful high throughput data analysis platform for Life Sciences with over 15,000 users worldwide, is hiring. [Read more...](/galaxy-is-hiring/)
About Galaxy
The GalaxyTeam is a part of BX at Penn State, and the Biology and Mathematics and Computer Science departments at Emory University.
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State, and Emory University.